Dermatofibrosarcoma Protuberans: Update on the Diagnosis and Treatment

Abstract
:1. Introduction
2. Epidemiology
3. Pathogenesis
4. Clinical Characteristics
5. Clinical and Imaging Evaluation
6. Pathologic diagnosis
7. Differential diagnosis
8. Clinical Staging System
9. Treatment
9.1. Treatment of Resectable DFSP
9.2. Treatment of FS-DFSP
9.3. Treatment of Unresectable/Metastatic DFSP
9.3.1. Radiation Therapy
9.3.2. Targeted Therapy
10. Prognosis and Surveillance
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hoffmann, E. Über das knollentreibende Fibrosarkomder Haut (Dermatofibrosarkoma protuberans). Dermatology 1925, 43, 1–28. [Google Scholar] [CrossRef]
- Dominguez-Malagon, H.; Valdez-Carrillo Mdel, C.; Cano-Valdez, A.M. Dermatofibroma and dermatofibrosarcoma protuberans: A comparative ultrastructural study. Ultrastruct. Pathol. 2006, 30, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, I.; Kariya, Y.; Okada, E.; Yasuda, M.; Matori, S.; Ishikawa, O.; Uezato, H.; Takahashi, K. A Novel Chromosomal Translocation Associated With COL1A2-PDGFB Gene Fusion in Dermatofibrosarcoma Protuberans: PDGF Expression as a New Diagnostic Tool. JAMA Dermatol. 2015, 151, 1330–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pack, G.T.; Tabah, E.J. Dermato-fibrosarcoma protuberans. A report of 39 cases. A.M.A. Arch. Surg. 1951, 62, 391–411. [Google Scholar] [CrossRef]
- McPeak, C.J.; Cruz, T.; Nicastri, A.D. Dermatofibrosarcoma protuberans: An analysis of 86 cases-five with metastasis. Ann. Surg. 1967, 166, 803–816. [Google Scholar] [CrossRef]
- Smola, M.G.; Soyer, H.P.; Scharnagl, E. Surgical treatment of dermatofibrosarcoma protuberans. A retrospective study of 20 cases with review of literature. Eur. J. Surg. Oncol. 1991, 17, 447–453. [Google Scholar]
- Criscione, V.D.; Weinstock, M.A. Descriptive epidemiology of dermatofibrosarcoma protuberans in the United States, 1973 to 2002. J. Am. Acad. Dermatol. 2007, 56, 968–973. [Google Scholar] [CrossRef]
- Kreicher, K.L.; Kurlander, D.E.; Gittleman, H.R.; Barnholtz-Sloan, J.S.; Bordeaux, J.S. Incidence and Survival of Primary Dermatofibrosarcoma Protuberans in the United States. Dermatol. Surg. 2016, 42, S24–S31. [Google Scholar] [CrossRef]
- Larbcharoensub, N.; Kayankarnnavee, J.; Sanpaphant, S.; Kiranantawat, K.; Wirojtananugoon, C.; Sirikulchayanonta, V. Clinicopathological features of dermatofibrosarcoma protuberans. Oncol. Lett. 2016, 11, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Lyu, A.; Wang, Q. Dermatofibrosarcoma protuberans: A clinical analysis. Oncol. Lett. 2018, 16, 1855–1862. [Google Scholar] [CrossRef] [PubMed]
- Valdivielso-Ramos, M.; Hernanz, J.M. Dermatofibrosarcoma protuberans in childhood. Actas Dermo-Sifiliogr. 2012, 103, 863–873. [Google Scholar] [CrossRef]
- Kornik, R.I.; Muchard, L.K.; Teng, J.M. Dermatofibrosarcoma protuberans in children: An update on the diagnosis and treatment. Pediatric Dermatol. 2012, 29, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Mentzel, T.; Beham, A.; Katenkamp, D.; Dei Tos, A.P.; Fletcher, C.D. Fibrosarcomatous (“high-grade”) dermatofibrosarcoma protuberans: Clinicopathologic and immunohistochemical study of a series of 41 cases with emphasis on prognostic significance. Am. J. Surg. Pathol. 1998, 22, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.P.; Pedeutour, F.; Sirvent, N.; Grosgeorge, J.; Minoletti, F.; Coindre, J.M.; Terrier-Lacombe, M.J.; Mandahl, N.; Craver, R.D.; Blin, N.; et al. Deregulation of the platelet-derived growth factor B-chain gene via fusion with collagen gene COL1A1 in dermatofibrosarcoma protuberans and giant-cell fibroblastoma. Nat. Genet 1997, 15, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Sirvent, N.; Maire, G.; Pedeutour, F. Genetics of dermatofibrosarcoma protuberans family of tumors: From ring chromosomes to tyrosine kinase inhibitor treatment. Genes Chromosom. Cancer 2003, 37, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, L.; Maire, G.; Guillot, B.; Joujoux, J.M.; Follana, P.; Simon, M.P.; Coindre, J.M.; Pedeutour, F. Complex t(5;8) involving the CSPG2 and PTK2B genes in a case of dermatofibrosarcoma protuberans without the COL1A1-PDGFB fusion. Virchows Arch. Int. J. Pathol. 2008, 452, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Takahira, T.; Oda, Y.; Tamiya, S.; Yamamoto, H.; Kawaguchi, K.; Kobayashi, C.; Oda, S.; Iwamoto, Y.; Tsuneyoshi, M. Microsatellite instability and p53 mutation associated with tumor progression in dermatofibrosarcoma protuberans. Hum. Pathol. 2004, 35, 240–245. [Google Scholar] [CrossRef]
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar] [CrossRef]
- Jones, A.V.; Cross, N.C. Oncogenic derivatives of platelet-derived growth factor receptors. Cell. Mol. Life Sci. 2004, 61, 2912–2923. [Google Scholar] [CrossRef] [PubMed]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genome Res. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiraki-Hotokebuchi, Y.; Yamada, Y.; Kohashi, K.; Yamamoto, H.; Endo, M.; Setsu, N.; Yuki, K.; Ito, T.; Iwamoto, Y.; Furue, M.; et al. Alteration of PDGFRbeta-Akt-mTOR pathway signaling in fibrosarcomatous transformation of dermatofibrosarcoma protuberans. Hum. Pathol. 2017, 67, 60–68. [Google Scholar] [CrossRef]
- Shah, K.K.; McHugh, J.B.; Folpe, A.L.; Patel, R.M. Dermatofibrosarcoma Protuberans of Distal Extremities and Acral Sites: A Clinicopathologic Analysis of 27 Cases. Am. J. Surg. Pathol. 2018, 42, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Takahira, T.; Oda, Y.; Tamiya, S.; Higaki, K.; Yamamoto, H.; Kobayashi, C.; Izumi, T.; Tateishi, N.; Iwamoto, Y.; Tsuneyoshi, M. Detection of COL1A1-PDGFB fusion transcripts and PDGFB/PDGFRB mRNA expression in dermatofibrosarcoma protuberans. Mod. Pathol. 2007, 20, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Kutzner, H.; Mentzel, T.; Palmedo, G.; Hantschke, M.; Rutten, A.; Paredes, B.E.; Scharer, L.; Guillen, C.S.; Requena, L. Plaque-like CD34-positive dermal fibroma (“medallion-like dermal dendrocyte hamartoma”): Clinicopathologic, immunohistochemical, and molecular analysis of 5 cases emphasizing its distinction from superficial, plaque-like dermatofibrosarcoma protuberans. Am. J. Surg. Pathol. 2010, 34, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Dickson, B.C.; Hornick, J.L.; Fletcher, C.D.M.; Demicco, E.G.; Howarth, D.J.; Swanson, D.; Zhang, L.; Sung, Y.S.; Antonescu, C.R. Dermatofibrosarcoma protuberans with a novel COL6A3-PDGFD fusion gene and apparent predilection for breast. Genes Chromosom. Cancer 2018, 57, 437–445. [Google Scholar] [CrossRef]
- Dadone-Montaudie, B.; Alberti, L.; Duc, A.; Delespaul, L.; Lesluyes, T.; Perot, G.; Lancon, A.; Paindavoine, S.; Di Mauro, I.; Blay, J.Y.; et al. Alternative PDGFD rearrangements in dermatofibrosarcomas protuberans without PDGFB fusions. Mod. Pathol. 2018, 31, 1683–1693. [Google Scholar] [CrossRef]
- Hisaoka, M.; Okamoto, S.; Morimitsu, Y.; Tsuji, S.; Hashimoto, H. Dermatofibrosarcoma protuberans with fibrosarcomatous areas. Molecular abnormalities of the p53 pathway in fibrosarcomatous transformation of dermatofibrosarcoma protuberans. Virchows Archiv 1998, 433, 323–329. [Google Scholar] [CrossRef]
- Yong, P.F.; Grosse-Kreul, D.; Maher, J.; Salisbury, J.R.; Ibrahim, M.A. Dermatofibrosarcoma protuberans in a patient with X-linked agammaglobulinaemia. J. Clin. Pathol. 2007, 60, 1162–1164. [Google Scholar] [CrossRef] [Green Version]
- Kesserwan, C.; Sokolic, R.; Cowen, E.W.; Garabedian, E.; Heselmeyer-Haddad, K.; Lee, C.C.; Pittaluga, S.; Ortiz, C.; Baird, K.; Lopez-Terrada, D.; et al. Multicentric dermatofibrosarcoma protuberans in patients with adenosine deaminase-deficient severe combined immune deficiency. J. Allergy Clin. Immunol. 2012, 129, 762–769.e761. [Google Scholar] [CrossRef] [Green Version]
- Duffy, R.; Liaqat, M.; Lawrence, N.; Manders, S. Dermatofibrosarcoma protuberans in a pediatric patient with ataxia telangiectasia syndrome. Pediatric Dermatol. 2019, 36, 400–401. [Google Scholar] [CrossRef]
- Sapadin, A.N.; Gelfand, J.M.; Howe, K.L.; Phelps, R.G.; Grand, D.; Rudikoff, D. Dermatofibrosarcoma protuberans in two patients with acquired immunodeficiency syndrome. Cutis 2000, 65, 85–88. [Google Scholar] [PubMed]
- Anderson, K.A.; Vidimos, A.T. Two primary dermatofibrosarcoma protuberans associated with different pregnancies in a single patient. Dermatol. Surg. 2012, 38, 1876–1878. [Google Scholar] [CrossRef] [PubMed]
- Hanabusa, M.; Kamo, R.; Harada, T.; Ishii, M. Dermatofibrosarcoma protuberans with atrophic appearance at early stage of the tumor. J. Dermatol. 2007, 34, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Makino, M.; Sasaoka, S.; Nakanishi, G.; Makino, E.; Fujimoto, W. Congenital atrophic dermatofibrosarcoma protuberans detected by COL1A1-PDGFB rearrangement. Diagn. Pathol. 2016, 11, 24. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.; Combemale, P.; Dupin, M.; Chouvet, B.; Kanitakis, J.; Bouyssou-Gauthier, M.L.; Dubreuil, G.; Claudy, A.; Grimand, P.S. The atrophic variant of dermatofibrosarcoma protuberans in childhood: A report of six cases. Br. J. Dermatol. 1998, 139, 719–725. [Google Scholar]
- Laske, J.; Sergon, M.; Mentzel, T.; Beissert, S.; Maschke, J. Congenital dermatofibrosarcoma protuberans clinically mimicking a melanocytic naevus treated with serial excisions. JEADV 2017, 31, e541–e542. [Google Scholar] [CrossRef]
- Wrotnowski, U.; Cooper, P.H.; Shmookler, B.M. Fibrosarcomatous change in dermatofibrosarcoma protuberans. Am. J. Surg. Pathol. 1988, 12, 287–293. [Google Scholar] [CrossRef]
- Wang, C.; Luo, Z.; Chen, J.; Zheng, B.; Zhang, R.; Chen, Y.; Shi, Y. Target therapy of unresectable or metastatic dermatofibrosarcoma protuberans with imatinib mesylate: An analysis on 22 Chinese patients. Medicine 2015, 94, e773. [Google Scholar] [CrossRef]
- Rutkowski, P.; Klimczak, A.; Lugowska, I.; Jagielska, B.; Wagrodzki, M.; Debiec-Rychter, M.; Pienkowska-Grela, B.; Switaj, T. Long-term results of treatment of advanced dermatofibrosarcoma protuberans (DFSP) with imatinib mesylate-The impact of fibrosarcomatous transformation. Eur. J. Surg. Oncol. (EJSO) 2017, 43, 1134–1141. [Google Scholar] [CrossRef]
- Chilukuri, D.S.; Premkumar, P.; Venkitaraman, B.; Soundararajan, J.C.B. Pancreatic metastasis of dermatofibrosarcoma protuberans: A rare case. BMJ Case Rep. 2020, 13. [Google Scholar] [CrossRef]
- Fiore, M.; Miceli, R.; Mussi, C.; Lo Vullo, S.; Mariani, L.; Lozza, L.; Collini, P.; Olmi, P.; Casali, P.G.; Gronchi, A. Dermatofibrosarcoma protuberans treated at a single institution: A surgical disease with a high cure rate. J. Clin. Oncol. 2005, 23, 7669–7675. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, C.; Xiang, B.; Chen, S.; Li, L.; Ji, Y. Clinical Features, Pathological Findings and Treatment of Recurrent Dermatofibrosarcoma Protuberans. J. Cancer 2017, 8, 1319–1323. [Google Scholar] [CrossRef] [Green Version]
- Bowne, W.B.; Antonescu, C.R.; Leung, D.H.; Katz, S.C.; Hawkins, W.G.; Woodruff, J.M.; Brennan, M.F.; Lewis, J.J. Dermatofibrosarcoma protuberans: A clinicopathologic analysis of patients treated and followed at a single institution. Cancer 2000, 88, 2711–2720. [Google Scholar] [CrossRef]
- Chang, C.K.; Jacobs, I.A.; Salti, G.I. Outcomes of surgery for dermatofibrosarcoma protuberans. Eur. J. Surg. Oncol. (EJSO) 2004, 30, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Gloster, H.M., Jr. Dermatofibrosarcoma protuberans. J. Am. Acad. Dermatol. 1996, 35, 355–374. [Google Scholar] [CrossRef]
- LeBlanc, J.; Chan, C.; Zedlitz, A. Dermatofibrosarcoma protuberans. Cutis 2017, 100, E6–E7. [Google Scholar]
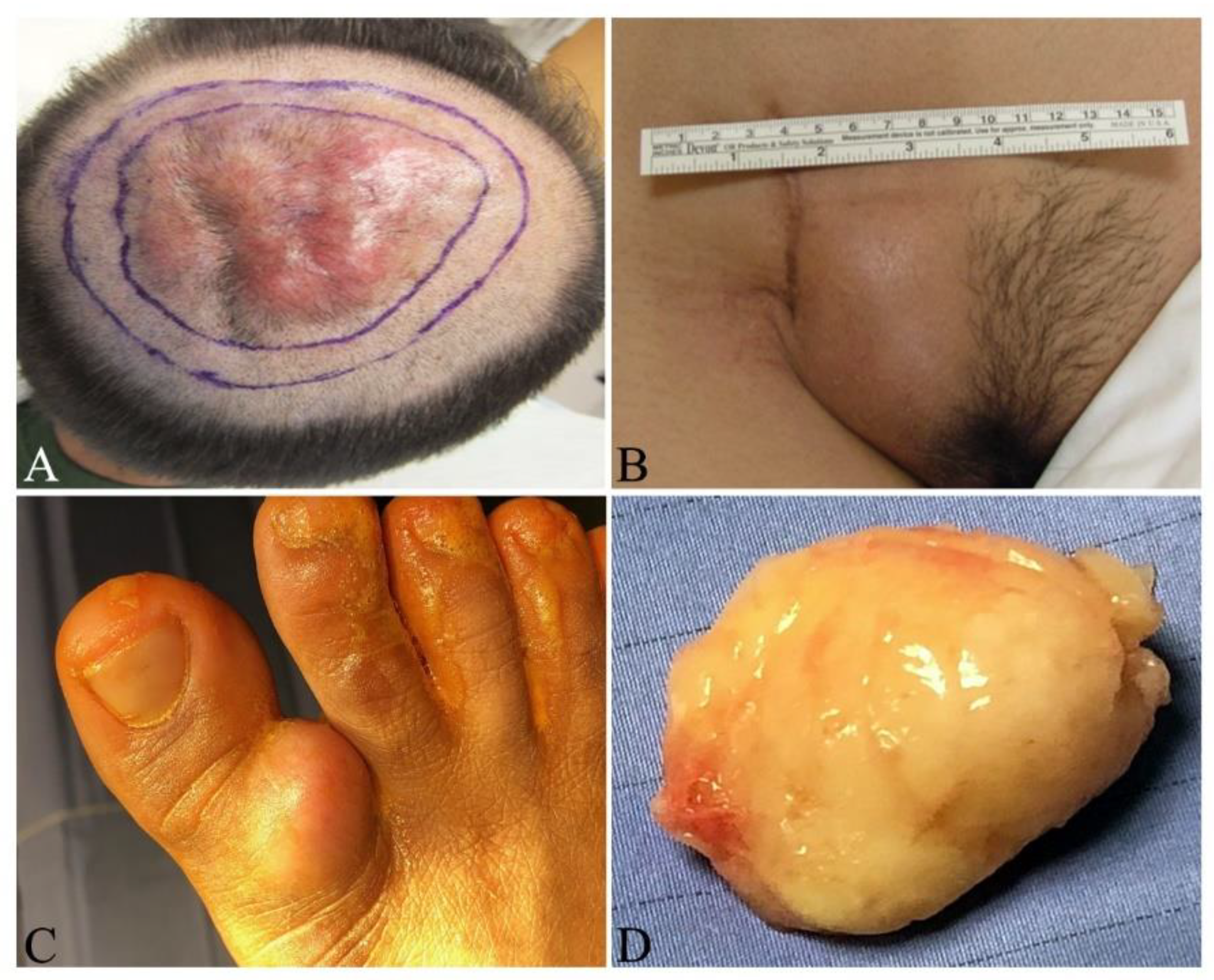
- Behfar, K.N.; Mendeszoon, M.J.; Chrzan, J.S.; Habershaw, G.M. Dermatofibrosarcoma protuberans of the hallux. J. Am. Acad. Dermatol. 1996, 86, 126–128. [Google Scholar] [CrossRef]
- Assassa, G.S.; Siegel, M.E.; Chen, D.C.; Ansari, A.N. Dermatofibrosarcoma protuberans of the toe. Findings on multiple imaging modalities. Clin. Nucl. Med. 1993, 18, 978–980. [Google Scholar] [CrossRef]
- Madden, C.; Spector, A.; Siddiqui, S.; Mirkin, G.; Yim, J.; Hao, X. Dermatofibrosarcoma Protuberans on Adult Toes: A Case Report and Review of the Literature. Anticancer. Res. 2019, 39, 2105–2111. [Google Scholar] [CrossRef]
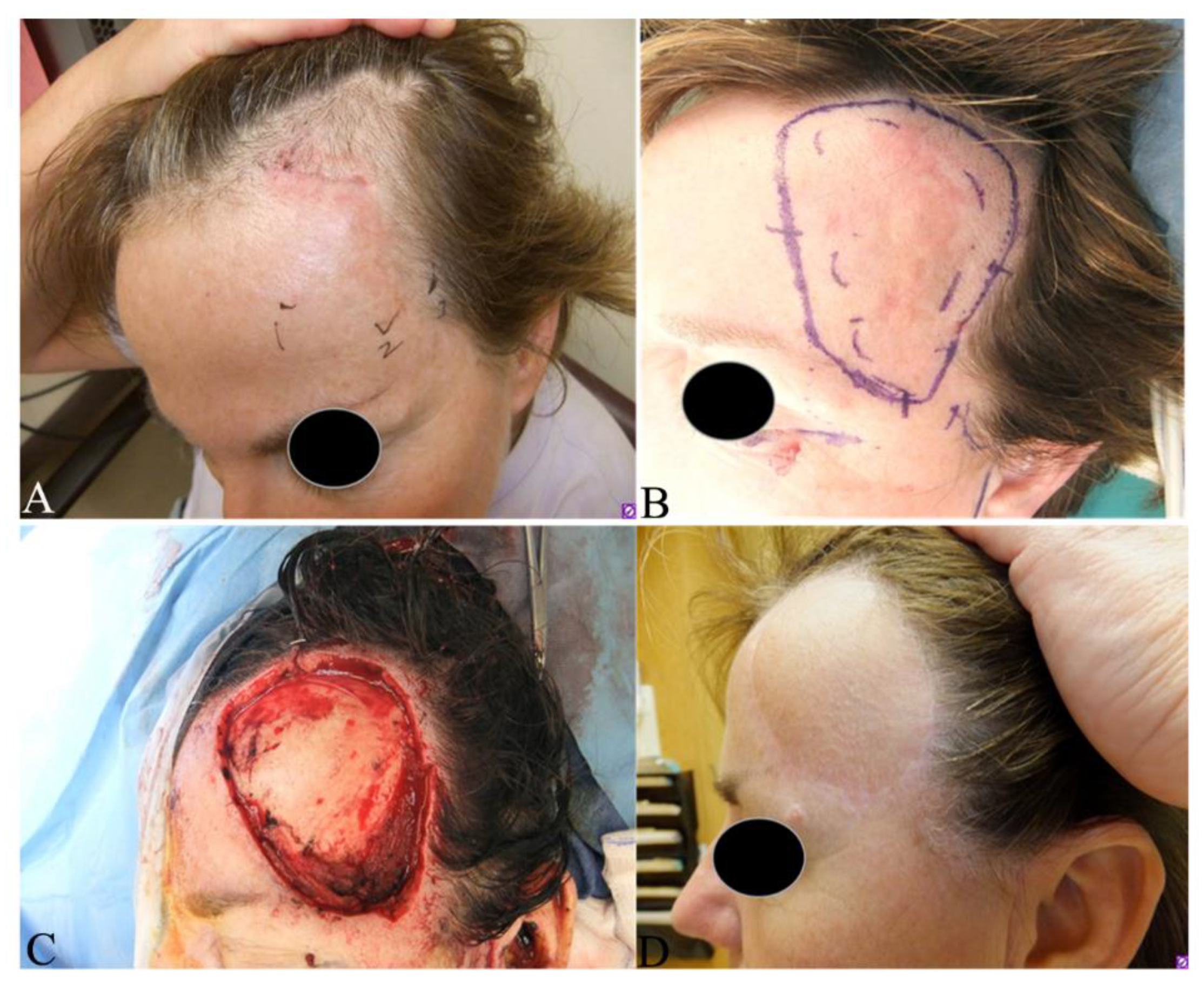
- Nelson, T.G.; Gonda, P.; Sheppard, P.; Keohane, S. Dermatofibrosarcoma Protuberans of the Scalp: A Challenging Tumor with a Proposed Modification to the Slow Mohs Technique. Dermatol. Surg. 2019. [Google Scholar] [CrossRef]
- Bouhani, M.; Fertani, Y.; Zemni, I.; Adouni, O.; Bouida, A.; Chargui, R.; Khaled, R. Dermatofibrosarcoma Protuberans of the Breast in Man: An Extremely Rare Entity With a Review of the Literature. J. Investig. Med. High Impact Case Rep. 2019, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecchio, G.M.; Broggi, G.; Mule, A.; Piombino, E.; Magro, G. Dermatofibrosarcoma protuberans: A tumor in the wide spectrum of the bland-looking spindle cell lesions of the breast. Pathologica 2019, 111, 87–91. [Google Scholar] [CrossRef] [Green Version]
- Kahn, T.A.; Liranzo, M.O.; Vidimos, A.T.; Papay, F.A.; Bergfeld, W.F. Pathological case of the month. Congenital dermatofibrosarcoma protuberans. Arch. Pediatr. Adolesc. Med. 1996, 150, 549–550. [Google Scholar] [CrossRef] [PubMed]
- Edelweiss, M.; Malpica, A. Dermatofibrosarcoma protuberans of the vulva: A clinicopathologic and immunohistochemical study of 13 cases. Am. J. Surg. Pathol. 2010, 34, 393–400. [Google Scholar] [CrossRef] [PubMed]
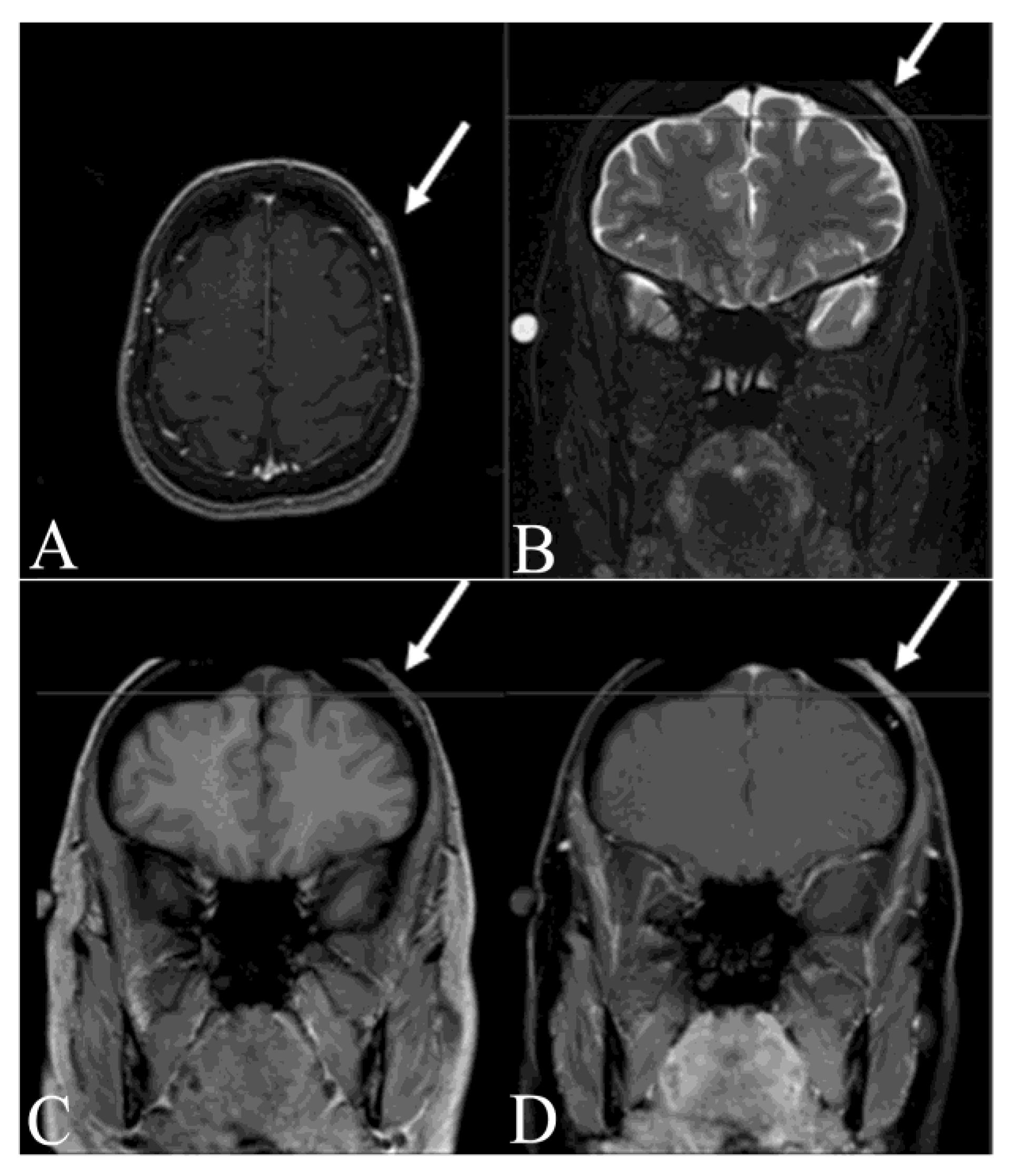
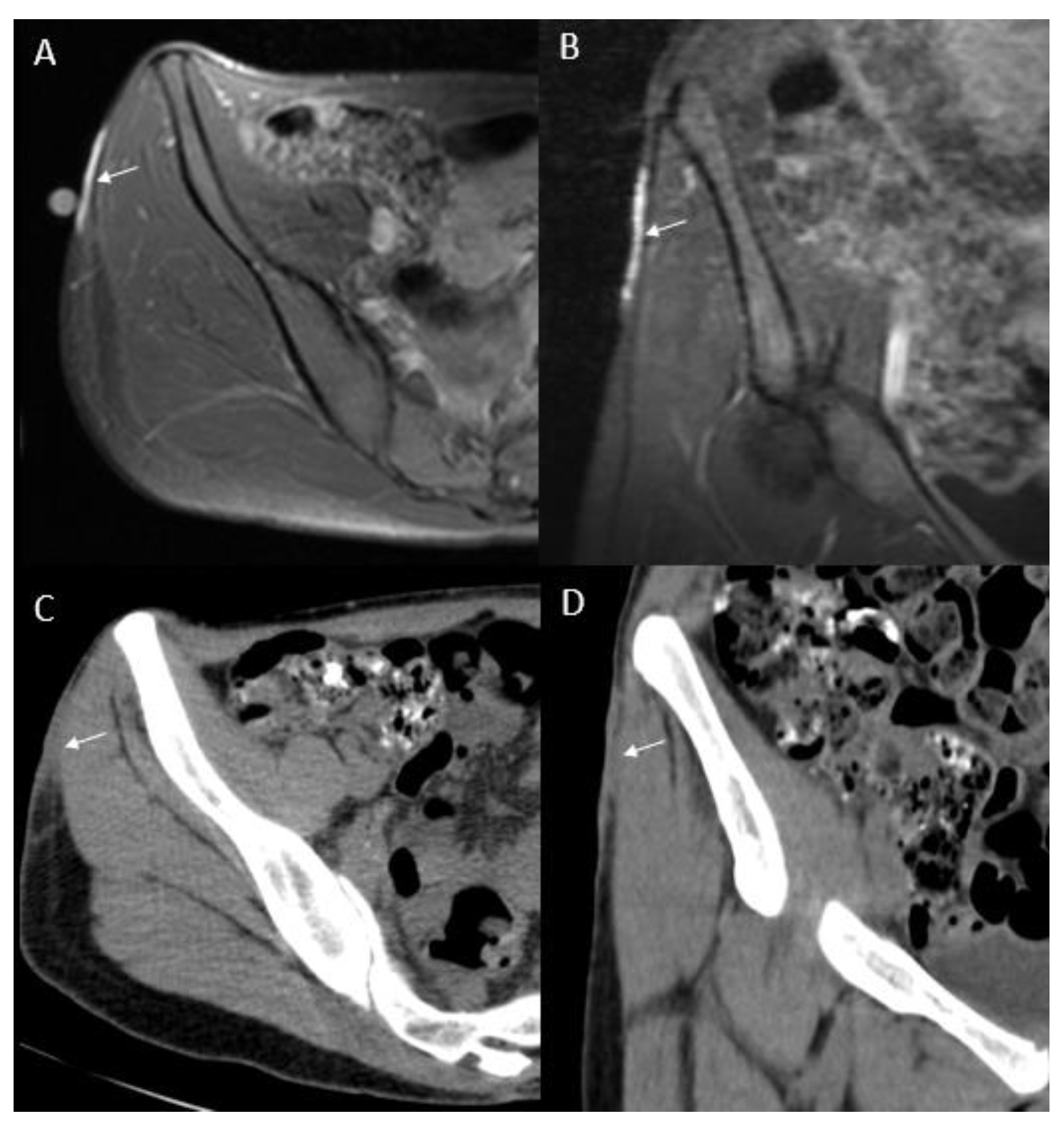
- Thornton, S.L.; Reid, J.; Papay, F.A.; Vidimos, A.T. Childhood dermatofibrosarcoma protuberans: Role of preoperative imaging. J. Am. Acad. Dermatol. 2005, 53, 76–83. [Google Scholar] [CrossRef]
- Riggs, K.; McGuigan, K.L.; Morrison, W.B.; Samie, F.H.; Humphreys, T. Role of magnetic resonance imaging in perioperative assessment of dermatofibrosarcoma protuberans. Dermatol. Surg. 2009, 35, 2036–2041. [Google Scholar] [CrossRef]
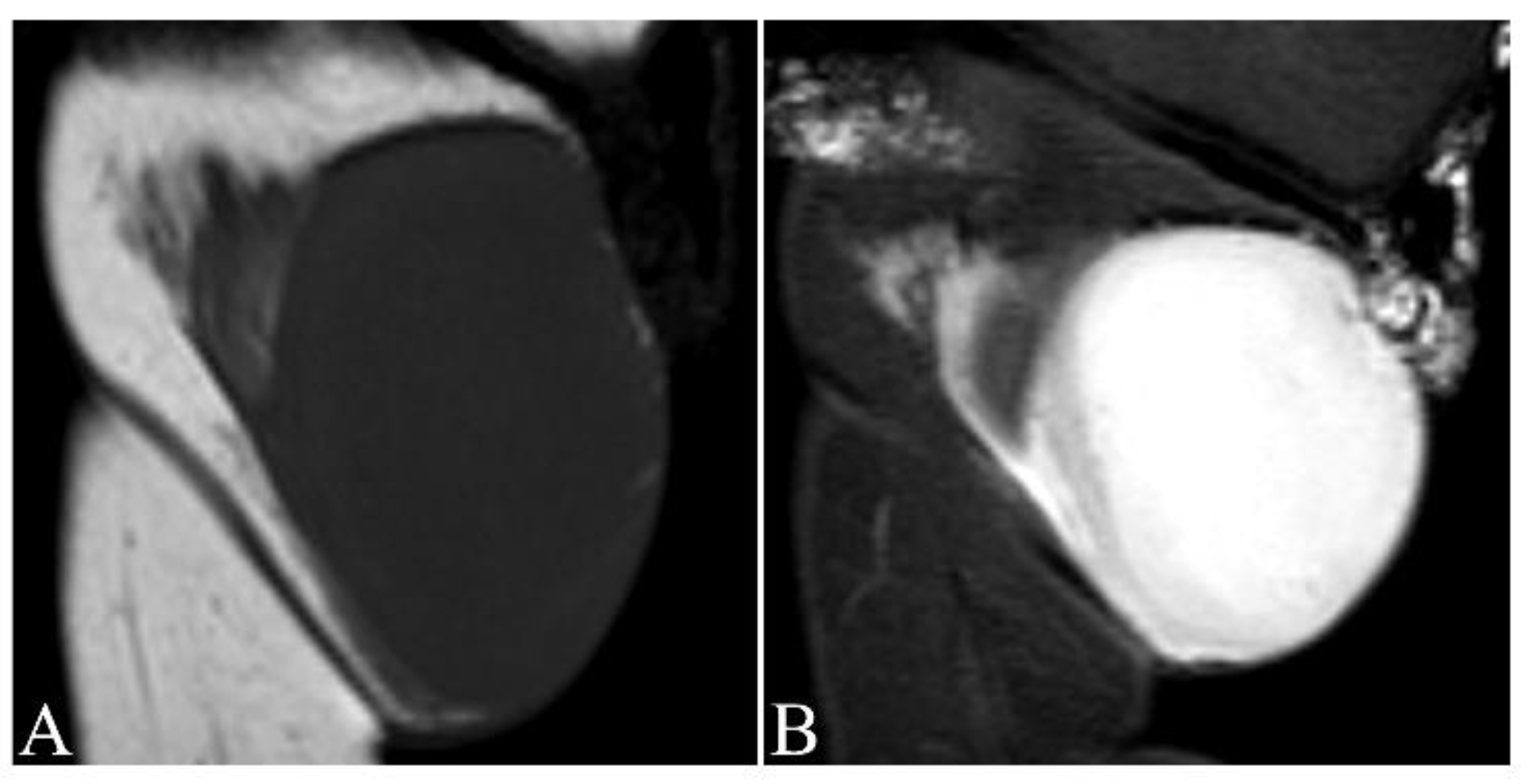
- Zhang, L.; Liu, Q.Y.; Cao, Y.; Zhong, J.S.; Zhang, W.D. Dermatofibrosarcoma Protuberans: Computed Tomography and Magnetic Resonance Imaging Findings. Medicine 2015, 94, e1001. [Google Scholar] [CrossRef]
- Torreggiani, W.C.; Al-Ismail, K.; Munk, P.L.; Nicolaou, S.; O’Connell, J.X.; Knowling, M.A. Dermatofibrosarcoma protuberans: MR imaging features. Am. J. Roentgenol. 2002, 178, 989–993. [Google Scholar] [CrossRef]
- Lee, M.H.; Kim, N.R.; Ryu, J.A. Cyst-like solid tumors of the musculoskeletal system: An analysis of ultrasound findings. Skelet. Radiol. 2010, 39, 981–986. [Google Scholar] [CrossRef]
- Rodriguez Bandera, A.I.; Moreno Bonilla, G.; Feito Rodriguez, M.; Beato Merino, M.J.; de Lucas Laguna, R. Jellyfish-like sonographic pattern can help recognition of dermatofibrosarcoma protuberans. Report of 3 new cases and review of the literature. Australas. J. Dermatol. 2019, 60, e148–e150. [Google Scholar] [CrossRef]
- Kransdorf, M.J.; Meis-Kindblom, J.M. Dermatofibrosarcoma protuberans: Radiologic appearance. Am. J. Roentgenol. 1994, 163, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, R.; Muddu, V.K.; Anantamakula, S.; Sri, S. Usefulness of (18)F-fluorodeoxyglucose positron emission tomography/computed tomography in dermatofibrosarcoma protuberans on treatment with imatinib. IJNM 2016, 31, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Suman, S.; Sharma, P.; Jain, T.K.; Sahoo, M.K.; Bal, C.; Kumar, R. Recurrent dermatofibrosarcoma protuberans with pulmonary metastases presenting twelve years after initial diagnosis: 18F-FDG PET/CT imaging findings. Clin. Nucl. Med. 2014, 39, 77–78. [Google Scholar] [CrossRef]
- Bague, S.; Folpe, A.L. Dermatofibrosarcoma protuberans presenting as a subcutaneous mass: A clinicopathological study of 15 cases with exclusive or near-exclusive subcutaneous involvement. Am. J. Dermatopathol. 2008, 30, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Llombart, B.; Monteagudo, C.; Sanmartin, O.; Lopez-Guerrero, J.A.; Serra-Guillen, C.; Poveda, A.; Jorda, E.; Fernandez-Serra, A.; Pellin, A.; Guillen, C.; et al. Dermatofibrosarcoma protuberans: A clinicopathological, immunohistochemical, genetic (COL1A1-PDGFB), and therapeutic study of low-grade versus high-grade (fibrosarcomatous) tumors. J. Am. Acad. Dermatol. 2011, 65, 564–575. [Google Scholar] [CrossRef]
- Liang, C.A.; Jambusaria-Pahlajani, A.; Karia, P.S.; Elenitsas, R.; Zhang, P.D.; Schmults, C.D. A systematic review of outcome data for dermatofibrosarcoma protuberans with and without fibrosarcomatous change. J. Am. Acad. Dermatol. 2014, 71, 781–786. [Google Scholar] [CrossRef]
- Lemm, D.; Mugge, L.O.; Mentzel, T.; Hoffken, K. Current treatment options in dermatofibrosarcoma protuberans. J. Cancer Res. Clin. Oncol. 2009, 135, 653–665. [Google Scholar] [CrossRef]
- Huis In ‘t Veld, E.A.; van Coevorden, F.; Grunhagen, D.J.; Smith, M.J.; van Akkooi, A.C.J.; Wouters, M.; Hayes, A.J.; Verhoef, C.; Strauss, D.C.; van Houdt, W.J. Outcome after surgical treatment of dermatofibrosarcoma protuberans: Is clinical follow-up always indicated? Cancer 2019, 125, 735–741. [Google Scholar] [CrossRef]
- Fletcher, C.D.; Theaker, J.M.; Flanagan, A.; Krausz, T. Pigmented dermatofibrosarcoma protuberans (Bednar tumour): Melanocytic colonization or neuroectodermal differentiation? A clinicopathological and immunohistochemical study. Histopathology 1988, 13, 631–643. [Google Scholar] [CrossRef]
- Terrier-Lacombe, M.J.; Guillou, L.; Maire, G.; Terrier, P.; Vince, D.R.; de Saint Aubain Somerhausen, N.; Collin, F.; Pedeutour, F.; Coindre, J.M. Dermatofibrosarcoma protuberans, giant cell fibroblastoma, and hybrid lesions in children: Clinicopathologic comparative analysis of 28 cases with molecular data--a study from the French Federation of Cancer Centers Sarcoma Group. Am. J. Surg. Pathol. 2003, 27, 27–39. [Google Scholar] [CrossRef]
- Jha, P.; Moosavi, C.; Fanburg-Smith, J.C. Giant cell fibroblastoma: An update and addition of 86 new cases from the Armed Forces Institute of Pathology, in honor of Dr. Franz, M. Enzinger. Ann. Diagn. Pathol. 2007, 11, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Craver, R.D.; Correa, H.; Kao, Y.S.; Van Brunt, T.; Golladay, E.S. Aggressive giant cell fibroblastoma with a balanced 17;22 translocation. Cancer Genet. Cytogenet. 1995, 80, 20–22. [Google Scholar] [CrossRef]
- Dal Cin, P.; Sciot, R.; de Wever, I.; Brock, P.; Casteels-Van Daele, M.; Van Damme, B.; Van Den Berghe, H. Cytogenetic and immunohistochemical evidence that giant cell fibroblastoma is related to dermatofibrosarcoma protuberans. Genes Chromosomes Cancer 1996, 15, 73–75. [Google Scholar] [CrossRef]
- O'Brien, K.P.; Seroussi, E.; Dal Cin, P.; Sciot, R.; Mandahl, N.; Fletcher, J.A.; Turc-Carel, C.; Dumanski, J.P. Various regions within the alpha-helical domain of the COL1A1 gene are fused to the second exon of the PDGFB gene in dermatofibrosarcomas and giant-cell fibroblastomas. Genes Chromosomes Cancer 1998, 23, 187–193. [Google Scholar]
- Harvell, J.D.; Kilpatrick, S.E.; White, W.L. Histogenetic relations between giant cell fibroblastoma and dermatofibrosarcoma protuberans. CD34 staining showing the spectrum and a simulator. Am. J. Dermatopathol. 1998, 20, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Braswell, D.S.; Ayoubi, N.; Motaparthi, K.; Walker, A. Dermatofibrosarcoma protuberans with features of giant cell fibroblastoma in an adult. J. Cutan. Pathol. 2020, 47, 317–320. [Google Scholar] [CrossRef]
- Maire, G.; Pedeutour, F.; Coindre, J.M. COL1A1-PDGFB gene fusion demonstrates a common histogenetic origin for dermatofibrosarcoma protuberans and its granular cell variant. Am. J. Surg. Pathol. 2002, 26, 932–937. [Google Scholar] [CrossRef]
- Sabater-Marco, V.; Perez-Valles, A.; Berzal-Cantalejo, F.; Rodriguez-Serna, M.; Martinez-Diaz, F.; Martorell-Cebollada, M. Sclerosing dermatofibrosarcoma protuberans (DFSP): An unusual variant with focus on the histopathologic differential diagnosis. Int. J. Dermatol. 2006, 45, 59–62. [Google Scholar] [CrossRef]
- Walluks, K.; Chen, Y.; Woelfel, C.; Yang, L.; Cui, T.; Seliger, C.; Geier, C.; Knosel, T.; Hauke, S.; Petersen, I. Molecular and clinicopathological analysis of dermatofibrosarcoma protuberans. Pathol. Res. Prac. 2013, 209, 30–35. [Google Scholar] [CrossRef]
- Han, T.Y.; Chang, H.S.; Lee, J.H.; Lee, W.M.; Son, S.J. A clinical and histopathological study of 122 cases of dermatofibroma (benign fibrous histiocytoma). Ann. Dermatol. 2011, 23, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Hao, X.; Levine, D.; Yim, J.; Qi, C.; Firestone, L.; Beiser, I.; Leone, E.; Woelffer, K.; Mirkin, G. Schwannoma of Foot and Ankle: Seven Case Reports and Literature Review. Anticancer Res. 2019, 39, 5185–5194. [Google Scholar] [CrossRef] [PubMed]
- Ortonne, N.; Wolkenstein, P.; Blakeley, J.O.; Korf, B.; Plotkin, S.R.; Riccardi, V.M.; Miller, D.C.; Huson, S.; Peltonen, J.; Rosenberg, A.; et al. Cutaneous neurofibromas: Current clinical and pathologic issues. Neurology 2018, 91, S5–S13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, A.; Tsuta, K.; Ohno, M.; Yoshida, M.; Narita, Y.; Kawai, A.; Asamura, H.; Kushima, R. STAT6 immunohistochemistry is helpful in the diagnosis of solitary fibrous tumors. Am. J. Surg. Pathol. 2014, 38, 552–559. [Google Scholar] [CrossRef]
- Sachdeva, M.P.; Goldblum, J.R.; Rubin, B.P.; Billings, S.D. Low-fat and fat-free pleomorphic lipomas: A diagnostic challenge. Am. J. Dermatopathol. 2009, 31, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, N.G.; Han, D. Desmoplastic melanoma. J. Surg. Oncol. 2019, 119, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Saiag, P.; Grob, J.J.; Lebbe, C.; Malvehy, J.; del Marmol, V.; Pehamberger, H.; Peris, K.; Stratigos, A.; Middelton, M.; Basholt, L.; et al. Diagnosis and treatment of dermatofibrosarcoma protuberans. European consensus-based interdisciplinary guideline. Eur. J. Cancer 2015, 51, 2604–2608. [Google Scholar] [CrossRef] [PubMed]
- Meguerditchian, A.N.; Wang, J.; Lema, B.; Kraybill, W.G.; Zeitouni, N.C.; Kane, J.M., 3rd. Wide excision or Mohs micrographic surgery for the treatment of primary dermatofibrosarcoma protuberans. Am. J. Clin. Oncol. 2010, 33, 300–303. [Google Scholar] [CrossRef]
- Paradisi, A.; Abeni, D.; Rusciani, A.; Cigna, E.; Wolter, M.; Scuderi, N.; Rusciani, L.; Kaufmann, R.; Podda, M. Dermatofibrosarcoma protuberans: Wide local excision vs. Mohs micrographic surgery. Cancer Treat. Rev. 2008, 34, 728–736. [Google Scholar] [CrossRef]
- Mullen, J.T. Dermatofibrosarcoma Protuberans: Wide Local Excision Versus Mohs Micrographic Surgery. Surg. Oncol. Clin. N. Am. 2016, 25, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Lowe, G.C.; Onajin, O.; Baum, C.L.; Otley, C.C.; Arpey, C.J.; Roenigk, R.K.; Brewer, J.D. A Comparison of Mohs Micrographic Surgery and Wide Local Excision for Treatment of Dermatofibrosarcoma Protuberans With Long-Term Follow-up: The Mayo Clinic Experience. Dermatol. Surg. 2017, 43, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Loghdey, M.S.; Varma, S.; Rajpara, S.M.; Al-Rawi, H.; Perks, G.; Perkins, W. Mohs micrographic surgery for dermatofibrosarcoma protuberans (DFSP): A single-centre series of 76 patients treated by frozen-section Mohs micrographic surgery with a review of the literature. J. Plast. Reconstr. Aesthetic Surg. 2014, 67, 1315–1321. [Google Scholar] [CrossRef]
- Malan, M.; Xuejingzi, W.; Quan, S.J. The efficacy of Mohs micrographic surgery over the traditional wide local excision surgery in the cure of dermatofibrosarcoma protuberans. Pan Afr. Med. J. 2019, 33, 297. [Google Scholar] [CrossRef] [PubMed]
- Foshee, J.P.; Trofymenko, O.; Zeitouni, N.C. Surgical and Functional Considerations of Dermatofibrosarcoma Protuberans Involving Facial Nerve Danger Zones. J. Clin. Aesthetic Dermatol. 2019, 12, 39–43. [Google Scholar]
- Schmults, C.D.; Rachael Blitzblau, R.; Engh, A. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) for Dermatofibrosarcoma Protuberans (Version 1.2020). © 2020 National Comprehensive Cancer Network, Inc.: Plymouth Meeting, PA, USA, 2019; Available online: https://linkprotect.cudasvc.com/url?a=https%3a%2f%2fnccn.org%2f.&c=E,1,lJrB7giSJWdXN-pBdcIkXYoOYTQT0hj26oGKyiktT0uURv6PjKeYKsXBoaccaaWH9_3DfJ4mTTuFEqjURd_xXSQeEa9Tv-WhbUrXhl5T7fw,&typo=1 (accessed on 1 June 2020).
- Ratner, D.; Thomas, C.O.; Johnson, T.M.; Sondak, V.K.; Hamilton, T.A.; Nelson, B.R.; Swanson, N.A.; Garcia, C.; Clark, R.E.; Grande, D.J. Mohs micrographic surgery for the treatment of dermatofibrosarcoma protuberans. Results of a multiinstitutional series with an analysis of the extent of microscopic spread. J. Am. Acad. Dermatol. 1997, 37, 600–613. [Google Scholar] [CrossRef]
- Monnier, D.; Vidal, C.; Martin, L.; Danzon, A.; Pelletier, F.; Puzenat, E.; Algros, M.P.; Blanc, D.; Laurent, R.; Humbert, P.H.; et al. Dermatofibrosarcoma protuberans: A population-based cancer registry descriptive study of 66 consecutive cases diagnosed between 1982 and 2002. J. Eur. Acad. Dermatol. Venereol. 2006, 20, 1237–1242. [Google Scholar] [CrossRef] [PubMed]
- Farma, J.M.; Ammori, J.B.; Zager, J.S.; Marzban, S.S.; Bui, M.M.; Bichakjian, C.K.; Johnson, T.M.; Lowe, L.; Sabel, M.S.; Wong, S.L.; et al. Dermatofibrosarcoma protuberans: How wide should we resect? Ann. Surg. Oncol. 2010, 17, 2112–2118. [Google Scholar] [CrossRef]
- Snow, H.; Davies, E.; Strauss, D.C.; Smith, M.; Hayes, A.J. Conservative Re-excision is a Safe and Simple Alternative to Radical Resection in Revision Surgery for Dermatofibrosarcoma Protuberans. Ann. Surg. Oncol. 2020, 27, 919–923. [Google Scholar] [CrossRef]
- Harati, K.; Lange, K.; Goertz, O.; Lahmer, A.; Kapalschinski, N.; Stricker, I.; Lehnhardt, M.; Daigeler, A. A single-institutional review of 68 patients with dermatofibrosarcoma protuberans: Wide re-excision after inadequate previous surgery results in a high rate of local control. World J. Surg. Oncol. 2017, 15, 5. [Google Scholar] [CrossRef] [Green Version]
- Abbott, J.J.; Oliveira, A.M.; Nascimento, A.G. The prognostic significance of fibrosarcomatous transformation in dermatofibrosarcoma protuberans. Am. J. Surg. Pathol. 2006, 30, 436–443. [Google Scholar] [CrossRef]
- Goldblum, J.R.; Reith, J.D.; Weiss, S.W. Sarcomas arising in dermatofibrosarcoma protuberans: A reappraisal of biologic behavior in eighteen cases treated by wide local excision with extended clinical follow up. Am. J. Surg. Pathol. 2000, 24, 1125–1130. [Google Scholar] [CrossRef]
- Williams, N.; Morris, C.G.; Kirwan, J.M.; Dagan, R.; Mendenhall, W.M. Radiotherapy for dermatofibrosarcoma protuberans. Am. J. Clin. Oncol. 2014, 37, 430–432. [Google Scholar] [CrossRef]
- Ballo, M.T.; Zagars, G.K.; Pisters, P.; Pollack, A. The role of radiation therapy in the management of dermatofibrosarcoma protuberans. Int. J. Radiat. Oncol. 1998, 40, 823–827. [Google Scholar] [CrossRef]
- Dagan, R.; Morris, C.G.; Zlotecki, R.A.; Scarborough, M.T.; Mendenhall, W.M. Radiotherapy in the treatment of dermatofibrosarcoma protuberans. Am. J. Clin. Oncol. 2005, 28, 537–539. [Google Scholar] [CrossRef]
- Castle, K.O.; Guadagnolo, B.A.; Tsai, C.J.; Feig, B.W.; Zagars, G.K. Dermatofibrosarcoma protuberans: Long-term outcomes of 53 patients treated with conservative surgery and radiation therapy. Int. J. Radiat. Oncol. 2013, 86, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Agyeman, M.B.; Vanderpuye, V.D.; Yarney, J. Abscopal Effect of Radiotherapy in Imatinib-resistant Dermatofibrosarcoma Protuberans. Cureus 2019, 11, e3857. [Google Scholar] [CrossRef] [Green Version]
- Ugurel, S.; Mentzel, T.; Utikal, J.; Helmbold, P.; Mohr, P.; Pfohler, C.; Schiller, M.; Hauschild, A.; Hein, R.; Kampgen, E.; et al. Neoadjuvant imatinib in advanced primary or locally recurrent dermatofibrosarcoma protuberans: A multicenter phase II DeCOG trial with long-term follow-up. Clin. Cancer Res. 2014, 20, 499–510. [Google Scholar] [CrossRef] [Green Version]
- Tazzari, M.; Indio, V.; Vergani, B.; De Cecco, L.; Rini, F.; Negri, T.; Camisaschi, C.; Fiore, M.; Stacchiotti, S.; Dagrada, G.P.; et al. Adaptive Immunity in Fibrosarcomatous Dermatofibrosarcoma Protuberans and Response to Imatinib Treatment. J. Investig. Dermatol. 2017, 137, 484–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McArthur, G.A. Molecular targeting of dermatofibrosarcoma protuberans: A new approach to a surgical disease. J. Natl. Compr. Cancer Netw. 2007, 5, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, S.; Pantaleo, M.A.; Negri, T.; Astolfi, A.; Tazzari, M.; Dagrada, G.P.; Urbini, M.; Indio, V.; Maestro, R.; Gronchi, A.; et al. Efficacy and Biological Activity of Imatinib in Metastatic Dermatofibrosarcoma Protuberans (DFSP). Clin. Cancer Res. 2016, 22, 837–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutkowski, P.; Van Glabbeke, M.; Rankin, C.J.; Ruka, W.; Rubin, B.P.; Debiec-Rychter, M.; Lazar, A.; Gelderblom, H.; Sciot, R.; Lopez-Terrada, D.; et al. Imatinib mesylate in advanced dermatofibrosarcoma protuberans: Pooled analysis of two phase II clinical trials. J. Clin. Oncol. 2010, 28, 1772–1779. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Dechent, C.; Mori, S.; Barker, C.A.; Dickson, M.A.; Nehal, K.S. Imatinib Treatment for Locally Advanced or Metastatic Dermatofibrosarcoma Protuberans: A Systematic Review. JAMA Dermatol. 2019, 155, 361–369. [Google Scholar] [CrossRef]
- Kerob, D.; Porcher, R.; Verola, O.; Dalle, S.; Maubec, E.; Aubin, F.; D’Incan, M.; Bodokh, I.; Boulinguez, S.; Madelaine-Chambrin, I.; et al. Imatinib mesylate as a preoperative therapy in dermatofibrosarcoma: Results of a multicenter phase II study on 25 patients. Clin. Cancer Res. 2010, 16, 3288–3295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Yin, Y.; Shen, C.; Yin, X.; Cai, Z.; Pu, L.; Fu, W.; Wang, Y.; Zhang, B. Preoperative imatinib treatment in patients with locally advanced and metastatic/recurrent gastrointestinal stromal tumors: A single-center analysis. Medicine 2020, 99, e19275. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, S.; Pedeutour, F.; Negri, T.; Conca, E.; Marrari, A.; Palassini, E.; Collini, P.; Keslair, F.; Morosi, C.; Gronchi, A.; et al. Dermatofibrosarcoma protuberans-derived fibrosarcoma: Clinical history, biological profile and sensitivity to imatinib. Int. J. Cancer 2011, 129, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- McArthur, G.A.; Demetri, G.D.; van Oosterom, A.; Heinrich, M.C.; Debiec-Rychter, M.; Corless, C.L.; Nikolova, Z.; Dimitrijevic, S.; Fletcher, J.A. Molecular and clinical analysis of locally advanced dermatofibrosarcoma protuberans treated with imatinib: Imatinib Target Exploration Consortium Study B2225. J. Clin. Oncol. 2005, 23, 866–873. [Google Scholar] [CrossRef]
- Kitagawa, D.; Yokota, K.; Gouda, M.; Narumi, Y.; Ohmoto, H.; Nishiwaki, E.; Akita, K.; Kirii, Y. Activity-based kinase profiling of approved tyrosine kinase inhibitors. Genes Cells 2013, 18, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.Y.; Liu, X.; Mao, M.; Li, M.; Choi, D.I.; Kang, S.W.; Lee, J.; La Choi, Y. Genetic aberrations in imatinib-resistant dermatofibrosarcoma protuberans revealed by whole genome sequencing. PLoS ONE 2013, 8, e69752. [Google Scholar] [CrossRef] [Green Version]
- Eilers, G.; Czaplinski, J.T.; Mayeda, M.; Bahri, N.; Tao, D.; Zhu, M.; Hornick, J.L.; Lindeman, N.I.; Sicinska, E.; Wagner, A.J.; et al. CDKN2A/p16 Loss Implicates CDK4 as a Therapeutic Target in Imatinib-Resistant Dermatofibrosarcoma Protuberans. Mol. Cancer Ther. 2015, 14, 1346–1353. [Google Scholar] [CrossRef] [Green Version]
- Oh, E.; Jeong, H.M.; Kwon, M.J.; Ha, S.Y.; Park, H.K.; Song, J.Y.; Kim, Y.J.; Choi, J.S.; Lee, E.H.; Lee, J.; et al. Unforeseen clonal evolution of tumor cell population in recurrent and metastatic dermatofibrosarcoma protuberans. PLoS ONE 2017, 12, e0185826. [Google Scholar] [CrossRef] [Green Version]
- Kamar, F.G.; Kairouz, V.F.; Sabri, A.N. Dermatofibrosarcoma protuberans (DFSP) successfully treated with sorafenib: Case report. Clin. Sarcoma Res. 2013, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyagawa, T.; Kadono, T.; Kimura, T.; Saigusa, R.; Yoshizaki, A.; Miyagaki, T.; Yamada, D.; Masui, Y.; Fujita, H.; Sato, S. Pazopanib induced a partial response in a patient with metastatic fibrosarcomatous dermatofibrosarcoma protuberans without genetic translocations resistant to mesna, doxorubicin, ifosfamide and dacarbazine chemotherapy and gemcitabine-docetaxel chemotherapy. J. Dermatol. 2017, 44, e21–e22. [Google Scholar] [CrossRef] [PubMed]
- Tsuchihashi, K.; Kusaba, H.; Yamada, Y.; Okumura, Y.; Shimokawa, H.; Komoda, M.; Uchino, K.; Yoshihiro, T.; Tsuruta, N.; Hanamura, F.; et al. Programmed death-ligand 1 expression is associated with fibrosarcomatous transformation of dermatofibrosarcoma protuberans. Mol. Clin. Oncol. 2017, 6, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Ohaegbulam, K.C.; Assal, A.; Lazar-Molnar, E.; Yao, Y.; Zang, X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol. Med. 2015, 21, 24–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
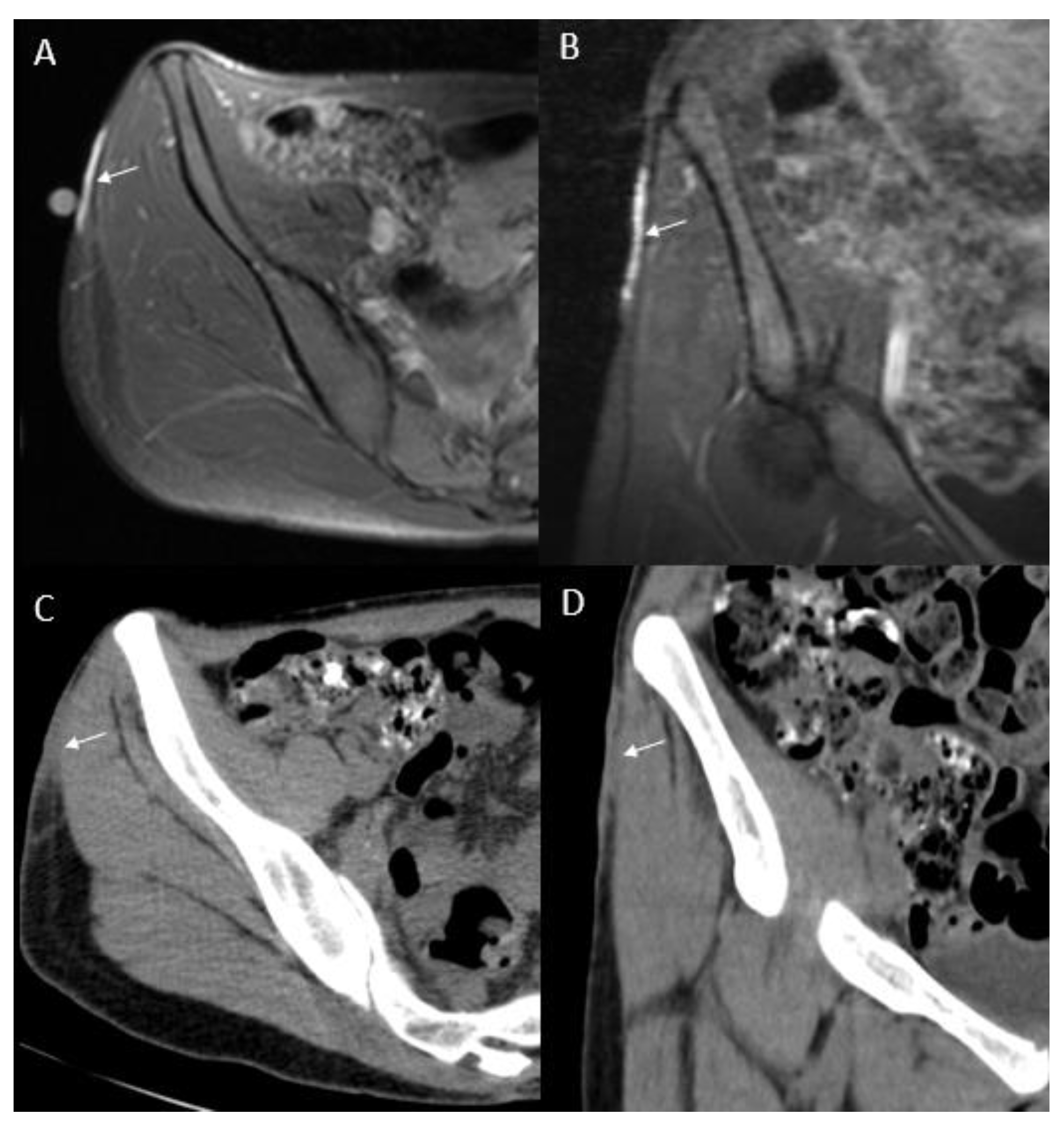


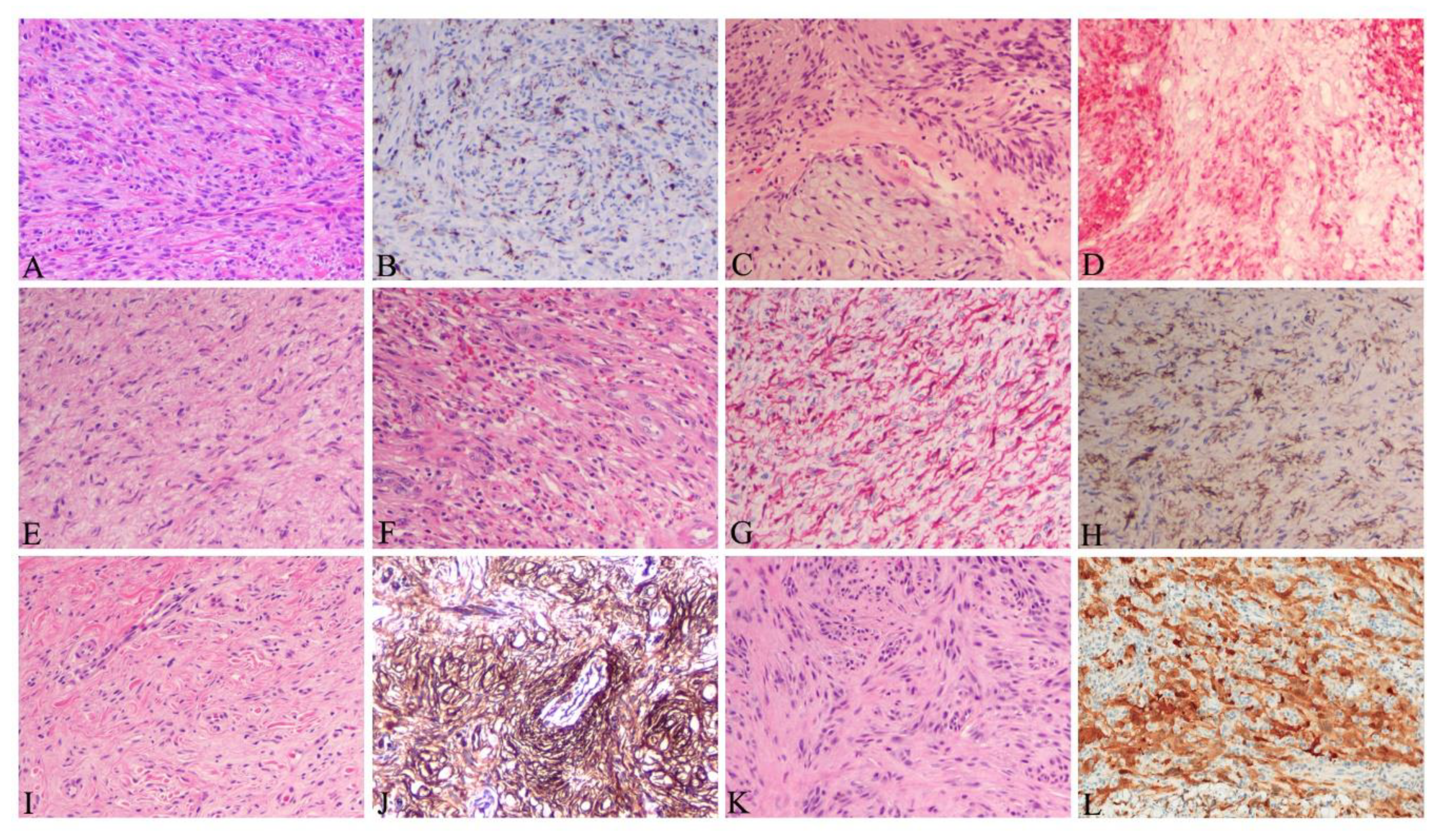
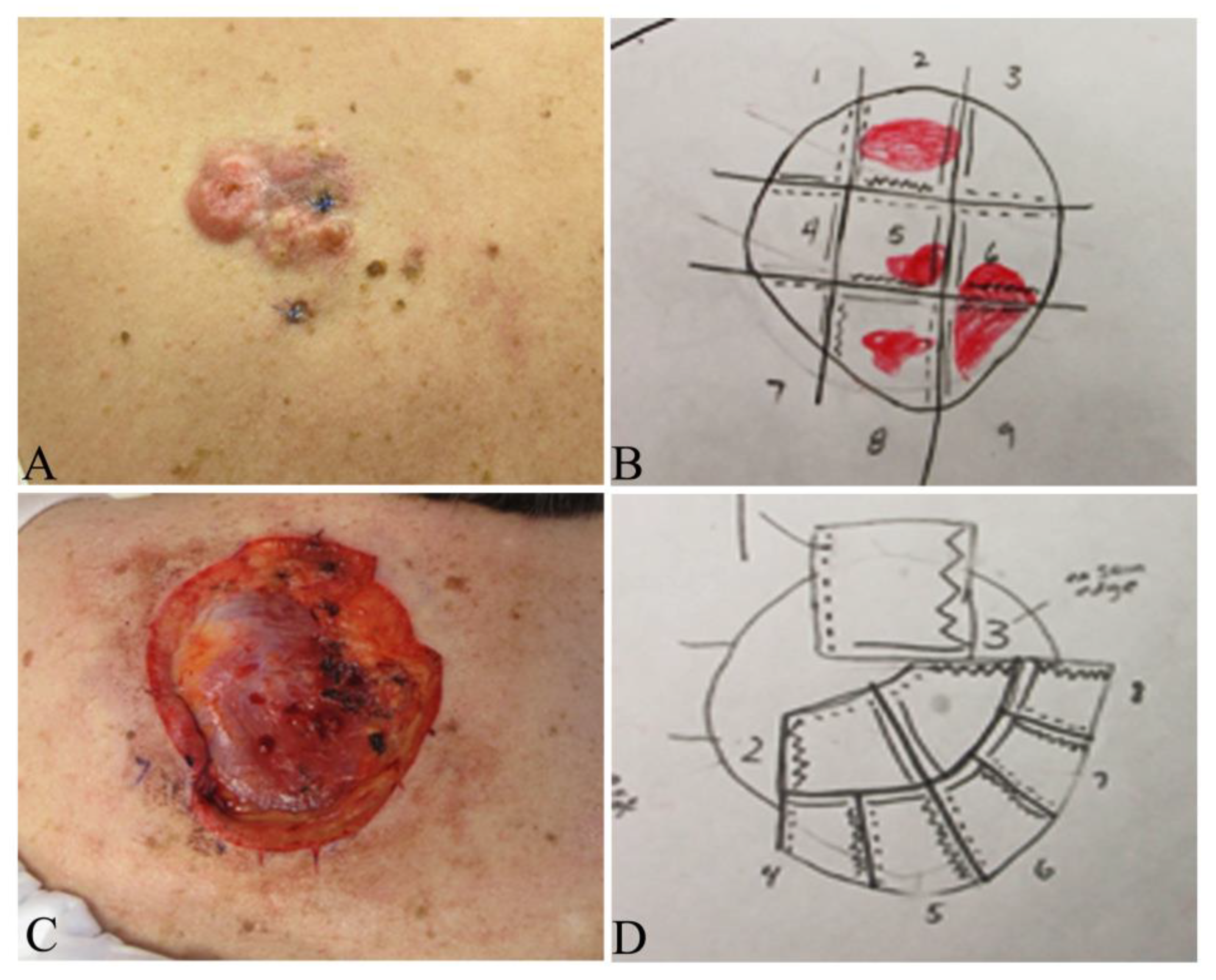

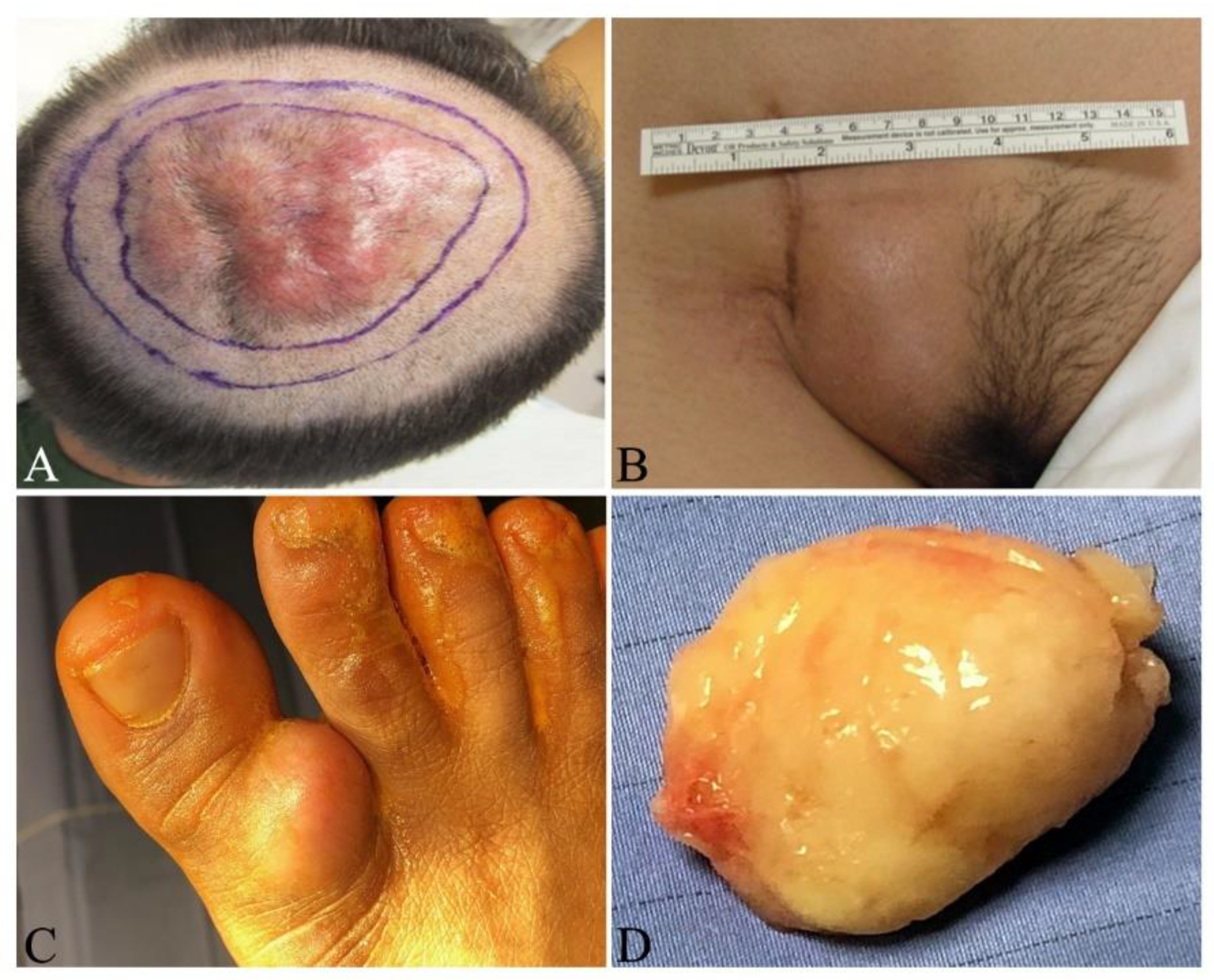
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
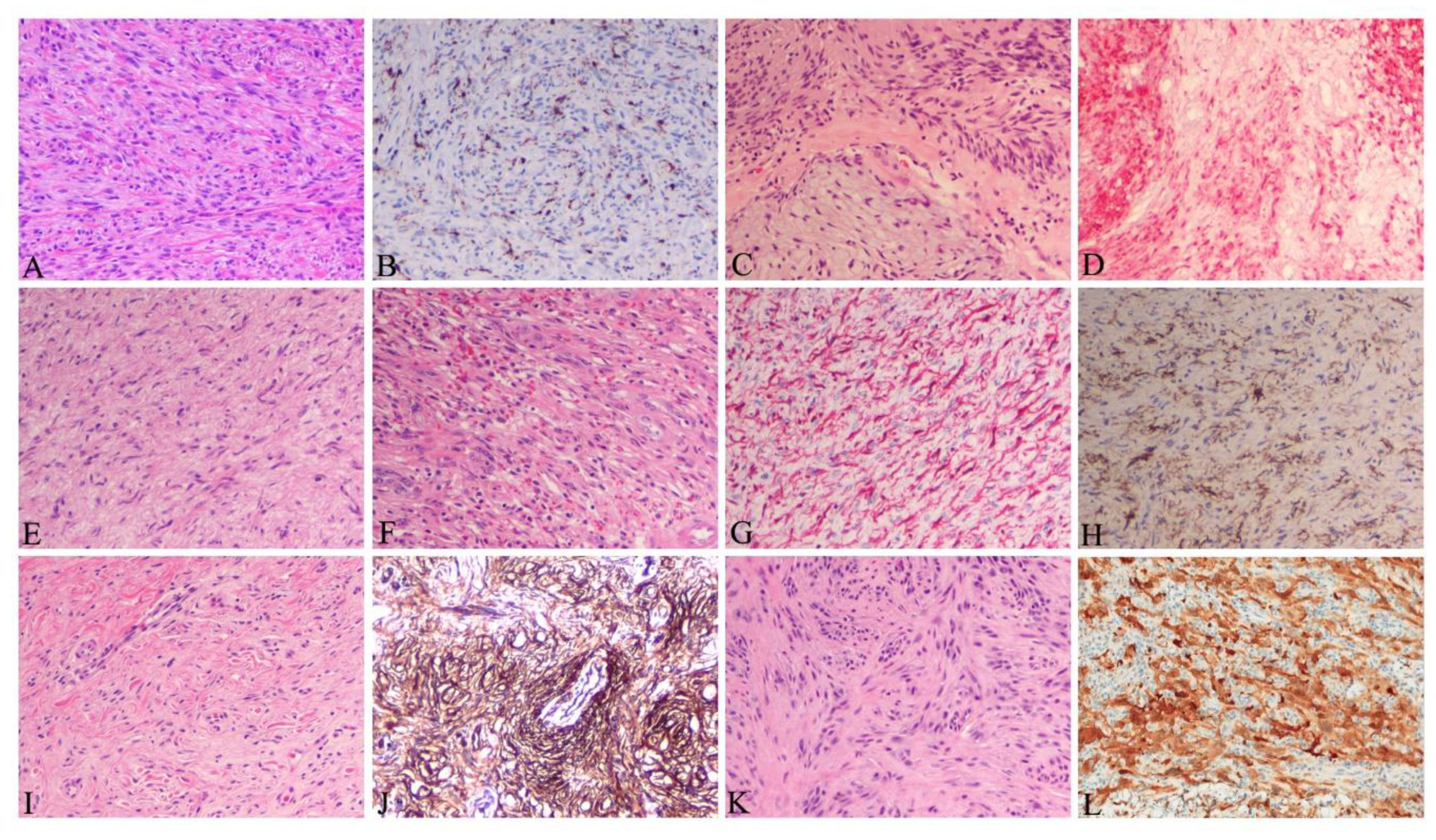
| Variants. | Histopathologic Features |
|---|---|
| Myxoid | Elongated, infiltrative spindle cells with myxoid changes in stroma (Figure 7E); CD34+, alpha smooth muscle actin-, desmin- |
| Pigmented (Bednář tumor) | Spindle cells admixed with scattered, single or a small cluster of dendritic melanin-containing cells [69]; CD34+; S-100+ and HBM45+ in pigmented cells |
| Giant cell (Rare) | Spindle cells admixed with pleomorphic or multinucleated giant cells (Figure 7F) [70,71]; CD34+ |
| giant cell fibroblastoma (GCF) (commonly seen in the pediatric population) | Parallel fascicles of wavy uniform spindled cells with wiry collagen, dense sclerosis and pseudovascular spaces with scattered and rimming pleomorphic giant cells [71]. GCF shares the same genetic abnormality as DFSP and the recurrent cases of GCF show histological features of DFSP [72,73,74,75,76] |
| Granular cell (Rare) | Spindle cells admixed with a proportion of cells with eccentric round nuclei, prominent nucleoli and abundant lysosomal granules [77]; CD34+, natural killer cell inhibitory factor 1C3+ |
| Sclerotic (Rare) | Spindle cells embedded in more than half of hypocellular collagenous components [78] (Figure 7G), CD34+ |
| Fibrosarcomatous (13.5%) [66] | Increased spindle cells with atypia, increased mitotic figures; fascicular or herringbone rather than storiform pattern; necrosis occasionally observed (Figure 7H) [66]; reduced or even lost CD34 expression (Figure 7I) [13] |
| Tumor | Clinical Feature | Histology | Immunostain |
|---|---|---|---|
| Dermatofibroma [80] | Elevated, pedunculated or dome shaped. More frequent in extremities, young (20–49 years) and females predominant | More pleomorphic with both small spindle-shaped fibroblastic cells and larger histiocytes admixed with chronic inflammatory cells in dermis (Figure 8A). Hyperkeratosis, acanthosis and pigmentation in epidermis. | XIIIa+ (Figure 8B) CD34- |
| Schwannoma [81] | Round, ovoid, well-circumscribed, solid mass, most common on the limbs, between 20–50 years | Well circumscribed with fibrous capsule, biphasic growth patterns with Antoni A (highly ordered wavy hyperchromatic spindle cells arranged in palisades) and with Antoni B (myxoid hypocellular components) (Figure 8C) | S100+(Figure 8D) |
| Cutaneous neurofibroma [82] | Skin colored, painless, slowly growing, solitary, soft, rubbery nodule. Frequently occurred in younger patients (20 to 40 years) | Mixed multiple cell types including Schwann cells, perineurial-like cells, fibroblastic cells, entrapped axons in interspersed with shredded carrot collagen, mast cells and lymphocytes (Figure 8E,F) | S-100+ (Figure 8G), Sox10+, CD34+ (Figure 8H), Collagen IV+, αSMA-, XIIIa- |
| Solitary fibrous tumor [83] | Usually occurred in older adults, slow-growing and painless mass with low rate of infiltration and metastasis | Relatively bland and uniform spindle cells within long, thin and parallel bands of collagen in “patternless” arrangement | CD34+, CD99+, STAT6+ [83], Vimentin+, Desmin-, S100- |
| Intradermal spindle cell lipoma [84] | Slowly growing, skin colored, raised, polypoid lesion with well-defined margin in seniors. | Bland spindle cells admixed with more or less or no matured lipocytes associated with delicate ropey/refractile collagen bundles (Figure 8I) | CD34+(Figure 8J), Rb-, S-100-, αSMA- |
| Spindle cell/ desmoplastic melanoma [85] | Pigmented or non-pigmented nodule on sun exposed skin in older adults | Significant atypia, pleomorphism, nuclear hyperchromasia, lack of storiform arrangement, pigmentations in spindle cells, derived from dysplastic melanocytic cells (Figure 8K) | S-100+(Figure 8L), typically Melan-A-and HMB-45-, sometimes, CD34+ |
| Stage | Criteria |
|---|---|
| Stage I | Non-protuberant lesions including atrophic or sclerotic plaque, macula or small nodules |
| Stage II | Protuberant primary tumor |
| Stage IIA | Superficial tumor: without invasion of the underlying fascia |
| Stage IIB | Deep tumor: either superficial to the fascia with infiltrating the fascia or occurred beneath the superficial fascia |
| Stage III | Lymph node metastasis |
| Stage IV | Distant metastasis to other organs |
| Comparable Parameter | WLE | MMS |
|---|---|---|
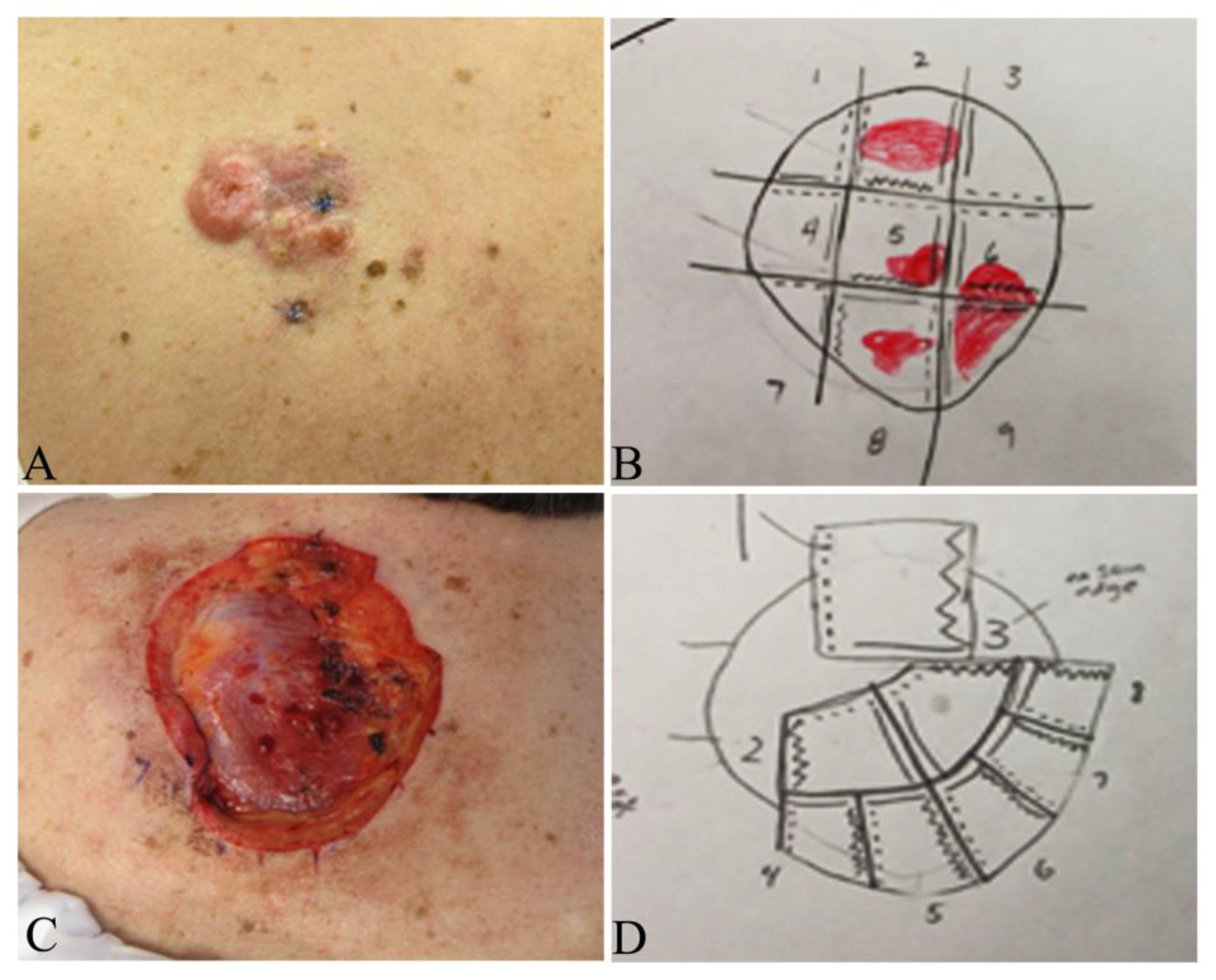
| Surgical procedures [87,88] | A three-dimensional excision including normal skin, subcutaneous tissue and the underlying investing fascia within a 2–4 cm margins from the gross tumor boundary | A stepwise procedure of tumor removal with mapping, histopathologic examination of 100% of the margins with tangential frozen sections by the Mohs surgeon and further deeper and/or wider re-excision of another layer of surrounding tissues if residual tumor cells are visualized. This procedure is repeated until all tumor margins are free of tumor cells. Usually performed as an outpatient with local anesthesia (Figure 9) |
| Advantages [87] | Relative simpler procedure Immediate wound repair following tumor removal Cost effective for patients and medical resources (but additional cost is incurred if positive margins need to be addressed) | Precise and complete evaluation of 100% of the surgical margins during excision; wound repair done when clear margins are obtained Lower rates of local recurrence compared with surgical excision |
| Drawbacks [87] | Unable to evaluate surgical margins during surgical operation Higher rate of local recurrences compared to Mohs surgery | Needs specialized training for Mohs surgeons and coordination with in office histotechnologists Delayed closure (usually same day) to allow for pathologic evaluation Time consuming and labor intensive May have higher cost for patients and medical resources |
| Applications [89] | Best for primary DFSPs on the trunk or extremities with a 2–4 cm margin from tumor boundary to completely excise tumor with acceptable cosmesis and function in a single operation | Ideal for DFSPs in cosmetically and functionally sensitive regions including face, scalp, neck, genitalia and digits to preserve tissue for optimal cosmetic reconstruction and functional recoveries and may be utilized in trunk and extremity DFSP |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, X.; Billings, S.D.; Wu, F.; Stultz, T.W.; Procop, G.W.; Mirkin, G.; Vidimos, A.T. Dermatofibrosarcoma Protuberans: Update on the Diagnosis and Treatment. J. Clin. Med. 2020, 9, 1752. https://doi.org/10.3390/jcm9061752
Hao X, Billings SD, Wu F, Stultz TW, Procop GW, Mirkin G, Vidimos AT. Dermatofibrosarcoma Protuberans: Update on the Diagnosis and Treatment. Journal of Clinical Medicine. 2020; 9(6):1752. https://doi.org/10.3390/jcm9061752
Chicago/Turabian StyleHao, Xingpei, Steven D. Billings, Fangbai Wu, Todd W. Stultz, Gary W. Procop, Gene Mirkin, and Allison T. Vidimos. 2020. "Dermatofibrosarcoma Protuberans: Update on the Diagnosis and Treatment" Journal of Clinical Medicine 9, no. 6: 1752. https://doi.org/10.3390/jcm9061752
APA StyleHao, X., Billings, S. D., Wu, F., Stultz, T. W., Procop, G. W., Mirkin, G., & Vidimos, A. T. (2020). Dermatofibrosarcoma Protuberans: Update on the Diagnosis and Treatment. Journal of Clinical Medicine, 9(6), 1752. https://doi.org/10.3390/jcm9061752